Nouveau règlement MDR relatifs aux dispositifs médicaux (UE 2017/745)

Ce que vous devez savoir sur le MDR
-
Le 5 avril 2017, le Parlement européen a adopté, en sus du règlement (UE) 2017/746 relatif aux dispositifs médicaux (DM) de diagnostic in vitro (RDIV), le nouveau règlement (UE) 2017/745 relatif aux dispositifs médicaux (RDM). Ces deux règlements qui sont entrés en vigueur le 26 mai 2017, sont d’application à compter du printemps 2021 (RDM) et du printemps 2022 (RDIV) avec différentes périodes transitoires échelonnées de six mois à cinq ans. Les deux nouveaux règlements de l’Union européenne (UE) relatifs aux dispositifs médicaux ont été adoptés par le Parlement européen le 5 avril 2017.
-
La réglementation de l’UE sur les dispositifs médicaux (Medical Devices Regulation, MDR) remplace les Directives MDD 93/42/CEE et AIMDD 90/385/CE.
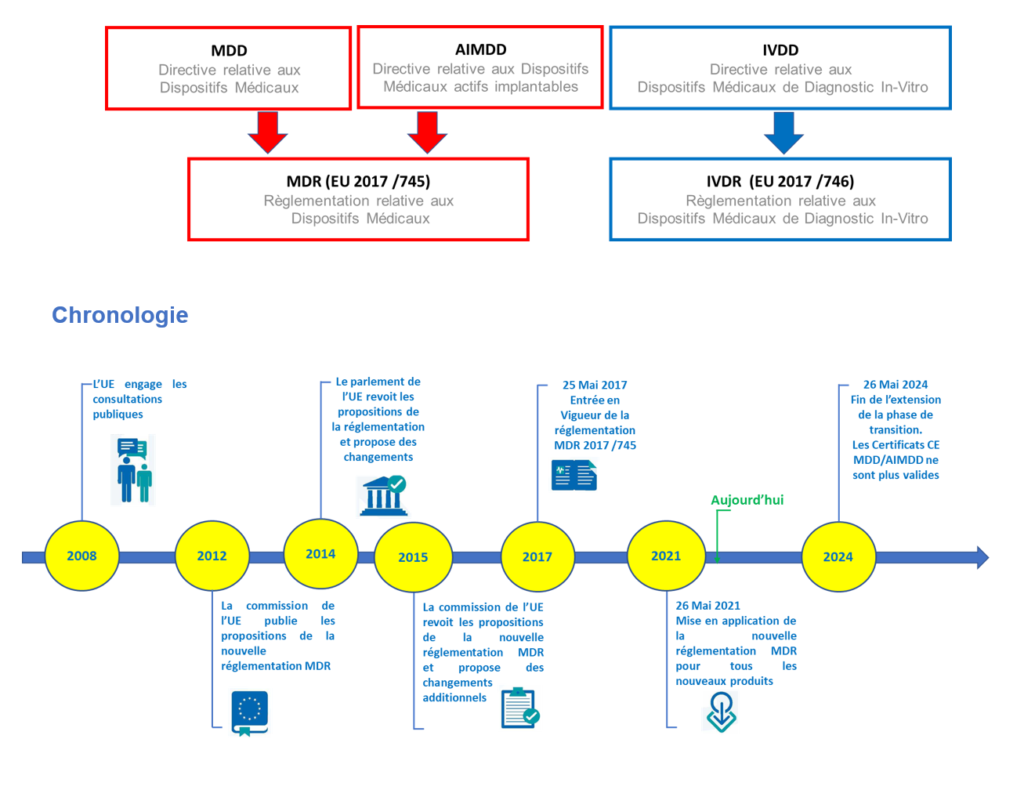
Les Changements Majeurs avec la nouvelle réglementation de l’UE 2017/745
1. Nouvelle Classification des dispositifs médicaux
-
La classe d’un DM est utilisée pour définir les exigences réglementaires applicables à un dispositif médical et aux activités de son fabricant, elle est directement liée à la dangerosité du dispositif.
-
En fonction du niveau de risque : I, IIa, IIb et III.
La classe I distingue les dispositifs intégrant une fonction de mesurage (Im) ainsi que les dispositifs stériles (Is) et les instruments chirurgicaux réutilisables (Icr).
-
L’utilisation prévue ou finalité (dispositif thérapeutique, de diagnostic, chirurgical), le caractère invasif (voire implantable), le caractère actif, la durée d’utilisation, les parties du corps concernées sont les facteurs déterminant la classe d’un dispositif médical.

2. Enregistrements du produit, des fabricants, distributeurs, importateurs sur la Plateforme Eudamed
3. Une personne en charge des aspects règlementaires
4. L’évaluation clinique est également à revoir, avec parfois de gros impacts
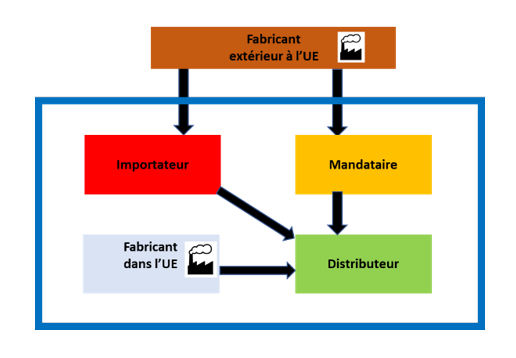
5. Le règlement clarifie les rôles et obligations des opérateurs économiques : Mandataire, Importateurs et Distributeurs.
6. La Surveillance Après Commercialisation (PMS – Post market surveillance)
7. Le Suivi Clinique Après Commercialisation
8. La nouvelle définition des spécifications communes et des standards harmonisés
9. Evaluation de la conformité du produit ( Conformity Assessment)
10. Déclaration des incidents et des FSCA (vigilance) 2, 10, 15 jours en fonction de leur criticité
11. Introduction et application de l’Identification Unique des produits (UDI -Chaque produit reçoit un numéro d’identification unique)
Situation de la Suisse
Le Conseil fédéral a adopté le 19 mai 2021 des dispositions venant compléter la législation d’exécution dans le domaine des dispositifs médicaux. Ces règles entrent en vigueur le 26 mai 2021, en même temps que l’ordonnance sur les dispositifs médicaux (ODim) entièrement révisée et la nouvelle ordonnance sur les essais cliniques de dispositifs médicaux (OClin-Dim).
La Suisse dispose depuis 2001 d’une réglementation sur les dispositifs médicaux équivalente à celle de l’UE et est liée par l’Accord de Reconnaissance Mutuel (ARM) au système de surveillance et au marché intérieur européen des dispositifs médicaux.
La Suisse a modifié ses bases légales, afin de maintenir l’équivalence entre le droit suisse et celui de l’UE. Sans mise à jour de l’ARM, les fabricants suisses doivent, à partir du 26 mai 2021, désigner un mandataire pour leurs produits dans l’UE.
La Suisse s’aligne sur le droit européen en ce qui concerne les dispositifs médicaux.
ARIAQ vous accompagne dans votre processus de mise à niveau
Il est capital de correctement planifier votre transition, La formation à cette nouvelle réglementation est déterminante. Faites-vous accompagner et commencez par identifier les changements. Contactez-nous pour en discuter. Au plaisir d’en parler avec Vous !
Max Ekobe – Formation et Conseil en dispositif médicaux
 Ingénieur diplômé en Management des projets Mécatroniques, Max Ekobe est spécialisé dans la fabrication de dispositifs médicaux stériles et de produits pharmaceutiques. Certifié BSI Lead Auditor ISO 13485. Max a plus de 15 ans d’expérience professionnelle au sein de fabricants internationaux de dispositifs médicaux stériles et de produits pharmaceutiques pour lesquels il a occupé des fonctions d’expert en validation des procédés de fabrication, de chef de projet, responsable ingénierie et responsable des opérations et Qualité. Il a durant sa carrière, géré des projets complexes et pluridisciplinaires.
Ingénieur diplômé en Management des projets Mécatroniques, Max Ekobe est spécialisé dans la fabrication de dispositifs médicaux stériles et de produits pharmaceutiques. Certifié BSI Lead Auditor ISO 13485. Max a plus de 15 ans d’expérience professionnelle au sein de fabricants internationaux de dispositifs médicaux stériles et de produits pharmaceutiques pour lesquels il a occupé des fonctions d’expert en validation des procédés de fabrication, de chef de projet, responsable ingénierie et responsable des opérations et Qualité. Il a durant sa carrière, géré des projets complexes et pluridisciplinaires.

 +41 24 423 96 50 | Horaires
+41 24 423 96 50 | Horaires